








CRISPR基因编辑解决方案
VectorBuilder可为体外和体内基因组编辑提供多种类型的CRISPR产品和服务。我们可提供包括质粒载体、病毒、IVT RNA和LNP、混合文库、细胞稳转株等多种CRISPR现货产品以满足多样的实验需求。
亮点

量身定制: 我们免费的、高度直观的线上载体设计平台可让您不受限制地设计您的CRISPR载体。

方案全面: 可提供多种基因递送方式,适用于基因敲除、敲入、以及使用Cas9变体的CRISPRa/i系统。

应用多样: 可提供从实验设计到细胞稳转株构建、文库筛选的全方位服务。

专业支持: 我们的产品服务质量优异、周期短、可提供具有竞争力的价格以及有力的技术支持。
我们提供的产品服务

CRISPR载体

CRISPR病毒

IVT RNA和LNP

Cas9蛋白

CRISPR CRO服务
基于我们免费的、用户友好的在线设计平台,您可以轻松设计和订购用于各种基因编辑和基因调控实验的定制或预制CRISPR载体。



VectorBuilder可提供极高质量的 病毒包装服务, 可实现对难转染细胞的高效CRISPR打靶。我们专有的病毒包装技术确保了包装的病毒具有更高的滴度、纯度、活性和一致性。



您可以使用我们的专有gRNA设计工具轻松设计gRNA,可保证高效且特异的基因打靶。您同时还可订购用于转染或显微注射的IVT gRNA和Cas9 mRNA。

VectorBuilder可提供为直接转染和高效编辑而优化的Cas9蛋白。




载体设计窍门
| 类型 | 建议 |
|---|---|
| CRISPR载体组件 |
更多信息请见此处。 |
| gRNA |
更多信息请见此处。 |
| Cas9 |
|
| PAM和gRNA的兼容性 |
|
| 报告基因系统 |
|
| 基因递送系统 |
更多信息请见此处。 |
| 供体DNA选择 |
更多信息请见此处。 |
| 靶位点 |
|
技术详情
CRISPR介导的基因组编辑
CRISPR介导的基因调控
CRISPR基因递送方法
gRNA数据库
CRISPR系统已在越来越多的实验中得到应用,每种CRISPR方法都至少由两个组分构成:一个Cas9蛋白以及向导RNA(gRNA)。最常用的Cas9来源于Streptococcus pyogenes细菌,是通过基因工程获得的(又名SpCas9,或着针对人类细胞进行密码子优化的hCas9)。它是一种 RNA 引导的 DNA 核酸酶,可以在靶位点产生DNA双链断裂 (DSB)(图1)。Cas9可通过与其互补配对的gRNA定位于宿主基因组的一个靶位点。当gRNA被设计用于靶向紧紧相邻原间隔区相邻基序(PAM)的区域时,就会引入DSB。PAM序列的选择取决于所使用的Cas酶类型:对于SpCas9而言,其对应的PAM序列为NGG或NAG(图1中的橙色DNA区域)。

图1 CRISPR引发的DNA修复产生基因敲除或精确序列变化的机制。
一旦通过CRISPR系统产生DSB,细胞通常会激活非同源末端连接 (NHEJ) 途径来修复DNA断裂,这通常会导致少量碱基的随机缺失,或者发生更罕见的碱基插入和碱基替换突变。当这些突变破坏了蛋白质编码区的序列时(例如发生移码突变),则可能引发功能性的基因敲除。双gRNA可以与Cas9一起引入以产生两个DSB,从而删除位于两个靶位点之间的DNA片段(图4),又或双gRNA可以用于同时靶向两个不同的基因。
另一种效率稍低的情况是,当引入DNA模板时,细胞可以通过同源重组修复 (HDR) 途径修复DSB。当供体DNA模板和CRISPR组件一同引入时,HDR可导致目标基因组DNA序列被供体序列替换,该机制有利于精确地改变DNA序列,例如产生点突变或在目标位点敲入供体DNA序列。供体DNA模板可以是单链寡核苷酸(ssODN)或dsDNA 片段(通常是来源于质粒或AAV的线性化DNA)。ssODN适用于引入点突变或小标签插入,而dsDNA片段则更适用于大片段敲入以实现转基因的靶向稳定整合。
另一种广泛使用的Cas9变体——Cas9“切口酶”(如Cas9(D10A)),能在DNA上产生单链切割。若将Cas9切口酶与gRNA结合使用,靶向同一目标区域两侧的两条互补链,切口酶会在两条链上各产生一个单链切割,致使目标区域周围形成大面积双链断裂(图2)。由于单链切口的修复具有高保真性,只有当两个错位的gRNA都正确结合时,才能实现高效切割并形成具有交错切口的DSB。因此,这种双切口策略在保持对多个位点高打靶效率的同时,能显著降低脱靶效应。

图2 切口酶结合2个gRNA后的酶切活性
失去催化活性的Cas9(dCas9)可以与不同形式的转录复合物结合以实现CRISPR介导的内源性基因转录调控。CRISPR介导的基因调控可用于多种应用,包括在染色质上测试调控元件、对目标细胞重编程,或用于全基因组筛选以识别促进增殖和分化的基因。
对于转录激活(CRISPRa),通常可使用协同激活介导系统(SAM)。该系统使用了三种组件:一个dCas9/VP64融合蛋白、一个MS2/P65/HSF1辅助蛋白复合体、以及一个经过修饰的gRNA(msgRNA)。dCas9蛋白经过改造结合了VP64,一种协同转录激活因子,而经过修饰的gRNA则含有一个MS2适配体,可以招募包含转录激活结构域NF-kB (p65)和HSF1的MS2复合物。总而言之,dCas9/VP64和MS2/P65/HSF1复合物协同作用,共同招募内源性的转录工具和辅助因子,从而激活靶基因表达,如图5所示。除了SAM系统,VectorBuilder还可以提供使用EB病毒R反式激活因子替换HSF1转录激活结构域的dCas9-VP64-p65-Rta(VPR)系统。已有报道表明,dCas9/VPR系统的内源性激活水平更高,但是dCas9/VP64由于较小的体积而更适合AAV载体系统。
相对的,如果需要抑制靶基因表达,dCas9也可以被改造为含有一个转录抑制结构域,如KRAB(krüppel-associated box)结构域和MeCP2。我们可提供两种dCas9/KRAB辅助载体:原始载体以及改造优化后的dCas9/KRAB/MeCP2载体。该改造版本将dCas9同时融合了两个转录抑制结构域KRAB/MeCP2,以便提供针对靶位点效果更强的转录抑制。KRAB结构域常在人的转录因子中被找到,是人类基因组中所发现的最常见的强效转录抑制因子之一。MeCP2可以通过招募组蛋白脱乙酰基酶,造成染色质凝聚和表观遗传抑制,进一步增强KRAB介导的转录抑制效果。当gRNA招募dCas9/KRAB/MeCP2到内源性启动子,如图6所示,转录被强力抑制。
CRISPR介导的基因组编辑或调控需要靶细胞同时表达Cas9和一个对靶位点特异的gRNA。无论应用何种方式,CRISPR组件必须被导入靶细胞。这些特定组件可通过以下不同途径进行转染或转导:
- gRNA和Cas9质粒
- gRNA和Cas9病毒(如慢病毒、AAV、腺病毒等)
- 混合的gRNA和Cas9 mRNA
- 由gRNA和Cas9蛋白组成的RNP复合物
图3总结了CRISPR基因递送的各种方法,图7、8、9和10中的案例研究展示了每种递送方法的有效性。
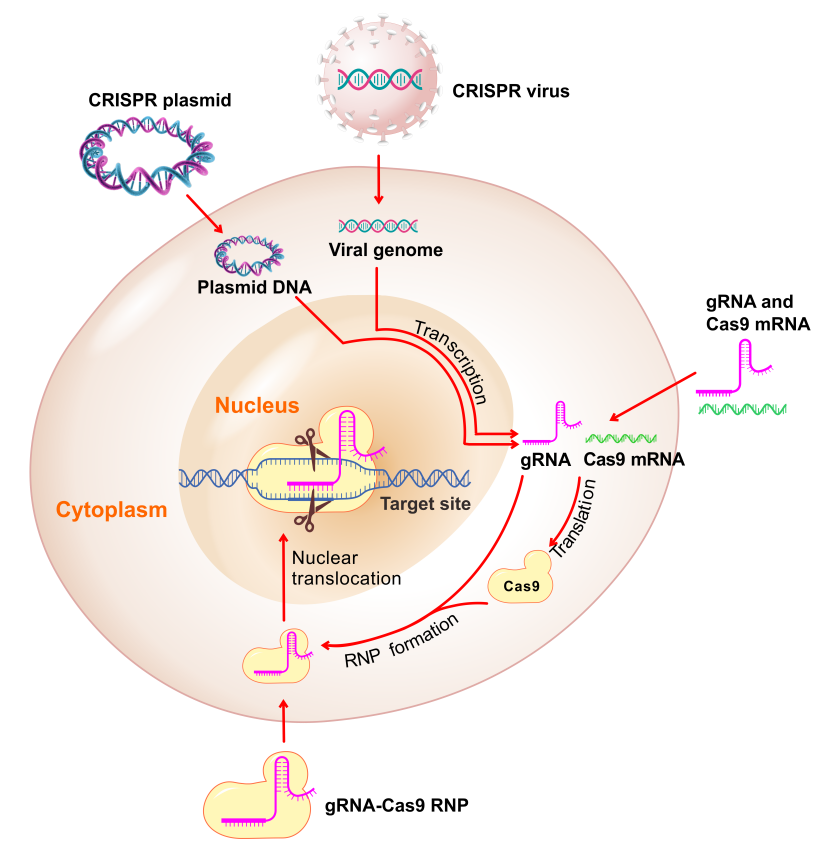
图3 将CRISPR组件递送到目标细胞的常用方法。
下表展示的是每种递送方式的优势与不足,可为您提供选择合适的CRISPR递送系统的相关参考:
| 递送方法 | 优势 | 不足 |
|---|---|---|
| gRNA与Cas9质粒 |
|
|
| gRNA与Cas9病毒 |
|
|
| gRNA和Cas9 mRNA的混合物 |
|
|
| 预制的的gRNA-Cas9 RNP复合物 |
|
|
VectorBuilder的线上CRISPR载体设计工具可提供针对人类、小鼠以及大鼠特征优化的全基因组gRNA数据库,旨在帮助您快速设计具有高打靶效率的CRISPR载体。我们仍在拓展我们的gRNA数据库,以囊括更多类型的Cas酶和更多的物种。我们的gRNA特异性评分计算遵循CRISPR文库设计(CLD)中的算法。简要而言,对于一个物种中的打靶N(20)NGG序列的一个给定的gRNA,我们搜索该物种基因组中的所有与靶序列比对≤3个错配碱基的潜在脱靶位点。以这种方式识别的每个潜在脱靶位点,都会计算一个脱靶分值。然后将所有脱靶位点的分值汇总用于计算gRNA的最终特异性分值,该数值介于0到100之间,值越高表示靶向特异性越强。请注意,特异性分值只是一个粗略的指导。实际的打靶效率和特异性可能与预测值不同。得分低的gRNA可能仍然有效。
当您在VectorBuilder的线上平台上设计CRISPR载体时,您可以在我们的数据库中搜索您的目标基因。输入基因名称后,您将在我们的数据库中看到针对您的目标基因的所有可用的gRNA的详细信息。
访问云舟学堂的教学资源,包括如何选择合适的CRISPR递送系统以及如何挑选恰当的CRISPR载体组件等相关指导,助力您成功规划、实施CRISPR实验,并解决实验中遇到的问题,从而确保实验取得成功。
案例研究
CRISPR介导的基因组编辑
CRISPR介导的基因调控
CRISPR递送方法

图4 使用gRNA/Cas9核糖核蛋白(RNP)方法,通过双gRNA删除DNA片段,生成纯合的靶基因敲除(KO)突变体。(A)编辑过的RNP复合物通过电转法导入靶细胞后可结合靶基因的两个位点以敲除一段长13 kb的区域,然后分离单克隆细胞并筛选。使用PCR和Sanger测序验证细胞的基因型。(B)在三个PCR反应中,使用了P1-P4四条引物以区分敲除和野生型克隆。(C)PCR结果验证了克隆1中的纯合敲除突变,并进一步在(D)靶基因测序结果中得到印证。

图5 利用基于慢病毒的CRISPRa系统上调基因的表达。向稳定表达dCas9/VP64和MS2/P65/HSF1 SAM复合物的NIH3T3细胞转导msgRNA表达慢病毒,然后进行药筛。(A)转录激活图示。(B)针对靶向小鼠mBrn2基因启动子区域而设计的msgRNA图示。(C)使用Scramble或打靶msgRNA分别转导NIH3T3细胞后与空白对照(NC)对比,用qRP-PCR测量Brn2基因的相对表达量。Mean±SD,*P<0.05,ANOVA结合Turkey事后检验。

图6 利用基于慢病毒的CRISPRi系统下调基因的表达。向稳定表达dCas9/KRAB/MeCP2的Jurkat 细胞转导表达gRNA的慢病毒,然后进行药筛。(A)转录抑制复合物图示。(B)靶向CXCR4基因启动子区的gRNA。(C)通过western blot显示了分别使用Scramble或gRNA转导的Jurkat细胞中以及空白对照组(NC)CXCR4蛋白质表达水平。(D)通过qRT-PCR测量CXCR4的相对基因表达。Mean±SD,***P<0.001,****P<0.0001,ANOVA之后进行Tukey事后检验。(E)流式细胞细胞术定量细胞表面表达的CXCR4。CXCR4使用单克隆一抗(Ab)和带有荧光基团的二抗来标记。未标记的和只标记二抗的Jurkat细胞均为阴性对照。(F)与转导scramble gRNA的细胞相比,转导了打靶CXCR4的gRNA的细胞其表面CXCR4数量减少了约50%。Mean±SD。
下列数种方法可用于将CRISPR组件递送至目的细胞中。每种方法均有其独特的优势和局限性。
- gRNA和Cas9质粒
- gRNA和Cas9病毒
- gRNA和Cas9 mRNA的混合物
- 含有gRNA和Cas9蛋白的RNP
质粒递送是将CRISPR组件引入细胞的最简单且最具成本效益的方法。

图7 All-in-one CRISPR系统介导的基因编辑。(A)向稳定表达EGFP的HEK293T细胞(HEK293T-EGFP)转染表达Cas9:T2A:mCherry,以及靶向EGFP的或scramble gRNA。转染后72小时,流式细胞术检测EGFP表达水平。MFI表示荧光强度平均值。(B)荧光显微镜下观察转导细胞中的EGFP表达情况(100X)。(C)实验组细胞对比无转染细胞中的相对EGFP表达。相对EGFP表达量用以下公式计算:[MFI (experimental group) – MFI (WT HEK293T cells)] / [MFI (HEK293T-EGFP cells) – MFI (WT HEK293T cells)]。Mean±SD,ns P>0.05,ANOVA结合Tukey事后检验。(D)gRNA靶向的基因组DNA区域使用PCR进行扩增,基因编辑效果使用T7E1试验确认。
慢病毒、AAV和腺病毒被广泛用于将CRISPR组件递送至哺乳动物细胞。

图8使用基于all-in-one方式设计的慢病毒CRISPR系统进行基因编辑。(A)将共表达Cas9:T2A:Puro与EGFP靶向gRNA或scramble gRNA的慢病毒载体包装成病毒颗粒,以MOI 10或50的比例转导稳定表达EGFP的HEK293T细胞。通过嘌呤霉素(Puro)筛选获得阳性转导细胞。(B)荧光显微镜下观察转导细胞中的EGFP表达情况(100X)。(C)从未转导细胞(NC)、scramble gRNA转导细胞、EGFP-1 gRNA转导细胞以及EGFP-2 gRNA转导细胞的基因组DNA中,PCR扩增gRNA打靶区域,并使用T7E1试验验证基因编辑效果。(D)通过流式细胞术定量EGFP阳性细胞。(E)使用MOI 10、50的慢病毒进行编辑后,EGFP阳性细胞比例分别对应降至28-32%、8-14%。***P<0.001表示gRNA(EGFP-1)与scramble gRNA比值,***P<0.001表示gRNA(EGFP-2)与scramble gRNA比值,ANOVA结合Dunnett事后检验。
gRNA和Cas9 mRNA的混合物提供了一种快速的基因组编辑方式,且没有对宿主基因组引入随机插入突变的风险。

图9 hSpCa9 IVT mRNA在体外的效果验证。(A)Cas9表达载体以T7启动子调控的方式设计,经体外转录(IVT)后的Cas9 mRNA与两种表达靶向EGFP 的gRNA共同转染至 HEK293T-EGFP细胞。(B)荧光显微镜下观察转导细胞中的EGFP表达情况(100X)。(C+D)流式细胞术定量检测荧光强度。转染Cas9和gRNA1或2后,相对EGFP表达量减少至大约40%-50%(D)。(E和F)从对照组和各实验组细胞的基因组DNA中分别通过PCR扩增出gRNA打靶区域。基因编辑效果通过T7E1酶切(E)和Sanger测序确认(F)。
纯化的 Cas9 蛋白与 gRNA 形成复合物,可提供一种精确、快速且高效的基因组编辑方法,尤其适用于需要瞬时活性的应用。

图10 CRISPR介导的iPSC中的基因敲入。通过电穿孔法将Cas9/gRNA核糖核蛋白复合体及供体载体导入iPSC,成功敲入UBC驱动的EGFP(2432 bp)。(A)通过Sanger测序验证EGFP在靶位点的敲入效果。(B)四个基因敲入纯合体单克隆的基因分型PCR检测。野生型靶位点区域长762 bp,敲入EGFP后靶位点区域长3194 bp。(C)显微镜下观察敲入细胞的EGFP荧光表达。(D)核型分析结果。(E)通过免疫荧光检测EGFP敲入的iPSC中NANOG、OCT4和SOX2等多能性标志物的表达,证实其细胞保持有多能性。
文档
暂无
常见问题解答
为了确定哪种方法最适合您的实验应用,以下是您应该考虑的一些事项。
基本原理
基因敲低载体
基因敲低载体表达shRNA抑制靶基因的mRNA,通过引发mRNA的切割抑制翻译过程。shRNA敲低不改变靶基因的DNA序列。
基因敲除载体
CRISPR和TALEN都是通过指导核酸酶切割基因组中的特定靶位点来发挥作用。然后这些切割位点被细胞低效地修复,从而导致修复部位的永久性突变,比如产生基因的小片段插入或碱基缺失。这类突变的一部分会导致基因的阅读框移码、出现提前终止密码子等,最终导致目的基因的功能丧失。如果同时靶向基因组中两个相邻紧密的切割位点(距离几个kb),也可以导致其间区域的删除。
效率
shRNA敲低永远不会完全抑制靶基因的表达。即使对于最有效的shRNA,靶基因的一些残留表达也会保留。相比之下,在一小部分经过处理的细胞群中,CRISPR和TALEN可以产生永久性突变,这可能导致基因功能完全丧失。
一致性和均一性
shRNA载体通常在转染/转导的细胞时获得较好的均一性,实验之间的一致性也较佳。相比之下,由于引入突变是随机的,CRISPR和TALEN的基因编辑效果在细胞之间的差异相当大。为了完全敲除细胞中的目的基因,必须敲除细胞中基因的所有拷贝。鉴于正常细胞具有任何基因的两个拷贝(X或Y连锁基因除外),而癌细胞可以具有两个以上的拷贝,这种完全敲除的细胞可能只占所有处理后的细胞的一小部分。出于这个原因,核酸酶介导的敲除实验需要通过测序来筛选克隆,以确定所有目的基因拷贝都被敲除的子细胞群。
脱靶效应
已有报道表明shRNA介导的基因敲低和核酸酶介导的基因敲除均会产生脱靶效应。脱靶效应的表征可以通过使用多个不同的shRNA 靶向同一基因来评估。如果一个基因被在多个不同的shRNA作用下仍然表现出一致的表征,则这种表征通常则不是脱靶效应带来的。对于CRISPR或TALEN基因敲除,应分析含有功能丧失突变的多个克隆,以包含可能由脱靶突变引起的任何表征。此外,可以对克隆进行脱靶位点测序,通过生物信息学方法鉴定以查看它们是否已发生突变。
CRISPR系统和TALEN系统均能用于在体外细胞和模式生物这种进行基因编辑。以下是这两种系统的特性对比:
基本原理
CRISPR
CRISPR系统使用位点特异的向导RNA (gRNA) 将Cas9核酸酶引导至其基因组中的目标位点以产生DNA切割。靶序列的长度通常约为20 bp。若靶序列中包含一些错配位点仍可能被识别和切割。
TALEN
TALEN系统使用的是一对嵌合蛋白,每个嵌合蛋白包含一个融合到Fokl核酸酶结构域的且作为TAL效应子的DNA结合结构域(识别特定DNA序列)。这对蛋白被设计成与基因组中的一对靶位点结合,每个靶位点长约18 bp,且二者相隔一个14-20 bp的间隔区。在与 DNA 结合后,这对蛋白上的Fokl核酸酶结构域发生二聚化,然后导致两个靶位点之间的间隔区内的序列发生切割。
效率
CRISPR和TALEN系统均在基因编辑上均表现出良好的效率,这也具体取决于应用的物种和细胞类型。一般来说,CRISPR系统的组分进入细胞后引发的DNA切割比TALEM系统更高效。
脱靶效应
CRISPR系统的gRNA靶向约20 bp的DNA序列,而TALEN系统需要约36 bp的靶序列。此外,Cas9/gRNA 复合物对靶序列中碱基错配(最多5 bp错配)的容忍度高于 TALEN。因此,TALEN介导的DNA切割比CRISPR具有更好的特异性,也不太可能在基因组中发生脱靶性的切割。相比之下,已有报道表明CRISPR系统在体外细胞系中可产生脱靶效应,而对CRISPR敲除小鼠的分析则表明体内实验时脱靶频率较低。最新的CRISPR系统的显着增强了CRISPR的特异性。通过使用双gRNA和Cas9切口酶(仅包含一个具有催化活性的核酸酶结构域的Cas9突变体,如Cas9(D10A)和Cas9_H840A),在靶向区域附近产生两个单链DNA切口,从而导致DSB发生在可修复的靶向区域内。在这种设计中由于双gRNA可将靶向序列的长度扩展至约40 bp,因此最大限度地减少了脱靶效应。
靶位点要求
TALEN系统可以针对基因组中的几乎任何位置进行设计。相比之下,CRISPR系统中的靶位点选择受限于PAM序列(通常为NGG)的要求,该序列位于gRNA靶向的DNA序列的3'末端。CRISPR系统的这种特性这并不会阻碍基因敲除,因为对靶基因中的任何地方切割都是有效的基因编辑,但可能难以对基因的特定位置进行切割或实现位点的特异性突变。为了通过将CRISPR系统应用于可精确编辑特定基因组位点,可以将包含所需编辑序列的同源重组供体载体或长寡核苷酸与靶位点的上游和下游同源臂一同递送至细胞中,以指导靶位点发生HDR介导的DNA修复。
技术难度
在基因编辑实验中,使用CRISPR系统在几个方面表现出对比TALEN具有更低的技术难道。 首先,对于载体构建,CRISPR 系统只需要合成一个短的gRNA,因为Cas9/gRNA 复合物的靶向依赖于机制简单的RNA/DNA杂交,而TALEN 系统需要根据每个独特的蛋白-DNA相互作用重新设计TAL的DNA结合域。与TALEN系统相比,gRNA显然是更便宜、更容易设计和构建的,而且TALEN系统打靶每个位点总是需要两个载体。其次,对于一些应用,例如注射小鼠胚胎,Cas9蛋白和gRNA可以通过直接注射来高效地递送,但TALEN系统不能。最后,CRISPR系统在基因筛选实验中的应用非常广泛,表达数千种不同gRNA的CRISPR筛选文库可以很容易地以高通量方式构建。
对于CRISPR介导的基因编辑,Cas9核酸酶通过特异性的gRNA作用至靶位点以产生DNA切割。在大多数情况下,为了实现简单的基因敲除,可以将单个gRNA与Cas9共同使用以产生DSB,然后通过NHEJ进行低效率的DNA修复,从而导致序列发生永久性突变,比如产生基因的小片段插入或碱基缺失。这类突变的一部分会导致基因的阅读框移码、出现提前终止密码子等,最终导致目的基因的功能丧失。
双gRNA则一般用于结合Cas9(D10A)切口酶对靶位点的两条反向DNA链进行切割。在这种方法中,切口酶将在两条链上产生单链切割,每一条链上的切割位点通过两条gRNA中的一条引导,然后在靶位点产生DSB。一般来说,这种方法减少潜在的脱靶效应,因为该种DSB的产生要求两条gRNA同时靶向序列。
当使用Cas9(D10A)切口酶和外源供体 DNA 模板将特定的碱基(例如敲入)引入感兴趣的基因时,也可以使用双gRNA。在这种方法中,两条反向的DNA链将被两个gRNA靶向位于所需突变位点两侧的两个位点,然后通过HDR途径利用外源DNA供体模板来修复切除的序列。
精选文献
Genetic and pharmacological modulation of DNA mismatch repair heterogeneous tumors promotes immune surveillance
Vito Amodio, et al.
Cancer Cell, 2023, doi: 10.1016/j.ccell.2022.12.003
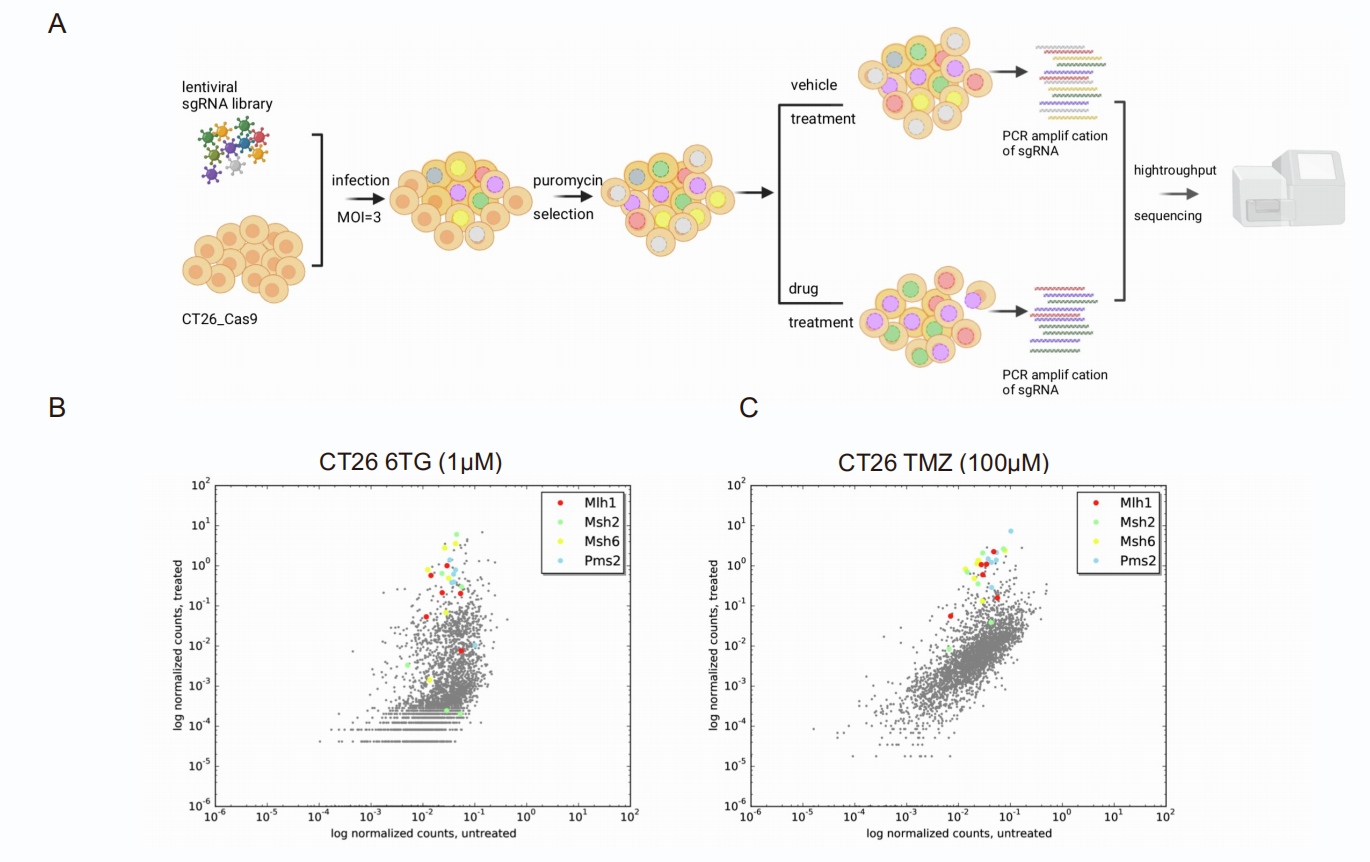
使用的云舟生物产品或服务:Abstract: Patients affected by colorectal cancer (CRC) with DNA mismatch repair deficiency (MMRd), often respond to immune checkpoint blockade therapies, while those with mismatch repair-proficient (MMRp) tumors generally do not. Interestingly, a subset of MMRp CRCs contains variable fractions of MMRd cells, but it is unknown how their presence impacts immune surveillance. We asked whether modulation of the MMRd fraction in MMR heterogeneous tumors acts as an endogenous cancer vaccine by promoting immune surveillance. To test this hypothesis, we use isogenic MMRp (Mlh1+/+) and MMRd (Mlh1-/-) mouse CRC cells. MMRp/MMRd cells mixed at different ratios are injected in immunocompetent mice and tumor rejection is observed when at least 50% of cells are MMRd. To enrich the MMRd fraction, MMRp/MMRd tumors are treated with 6-thioguanine, which leads to tumor rejection. These results suggest that genetic and pharmacological modulation of the DNA mismatch repair machinery potentiate the immunogenicity of MMR heterogeneous tumors.
>展开<收起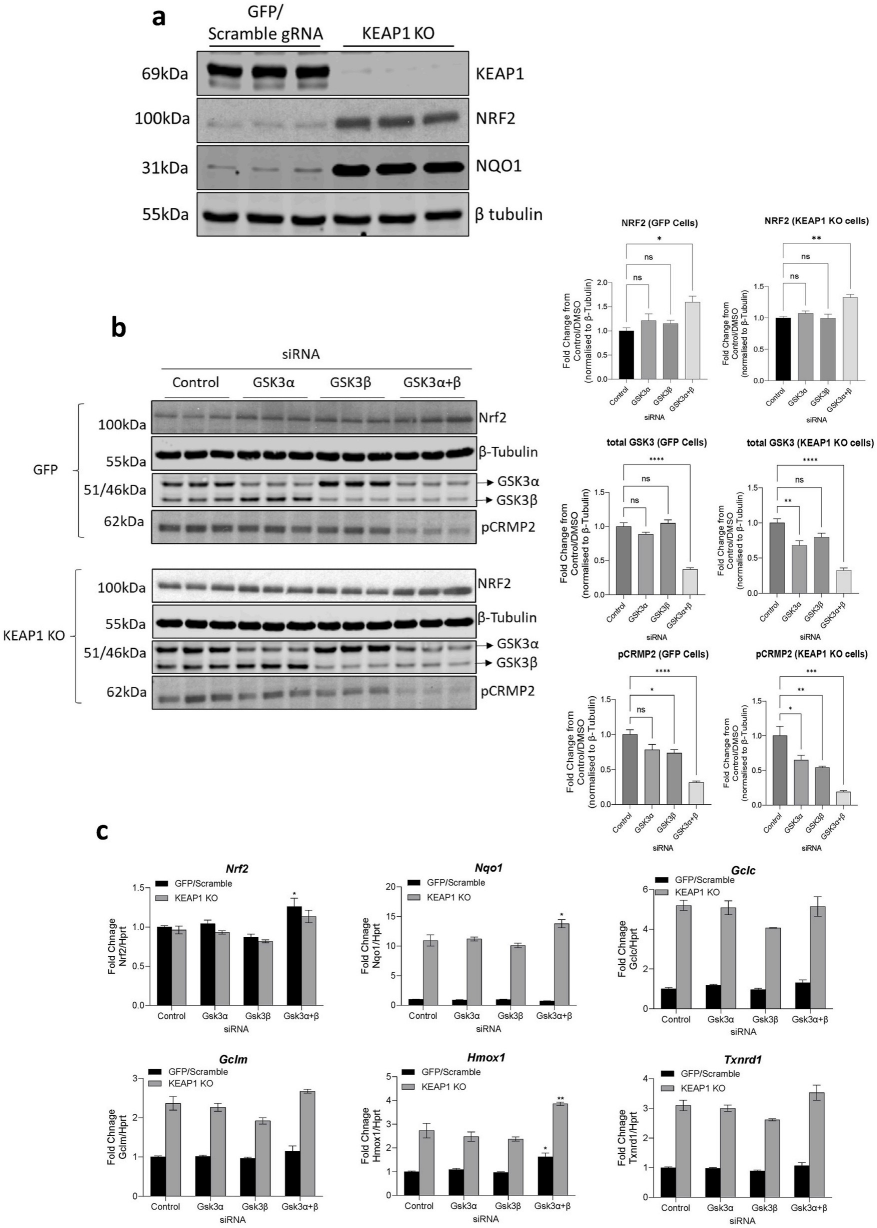
Inhibition of glycogen synthase kinase-3 enhances NRF2 protein stability, nuclear localisation and target gene transcription in pancreatic beta cells
Patibandla, et al.
Redox Biology, 2024, doi: 10.1016/j.redox.2024.103117
使用的云舟生物产品或服务:- Vector Cloning ( Mammalian CRISPR Lentiviral Vector )
- Vector Cloning ( Mammalian Gene Expression Adenoviral Vector )
- Lentivirus Packaging
- Adenovirus Packaging
Abstract: Accumulation of reactive oxygen species (i.e., oxidative stress) is a leading cause of beta cell dysfunction and apoptosis in diabetes. NRF2 (NF-E2 p45-related factor-2) regulates the adaptation to oxidative stress, and its activity is negatively regulated by the redox-sensitive CUL3 (cullin-3) ubiquitin ligase substrate adaptor KEAP1 (Kelch-like ECH-associated protein-1). Additionally, NRF2 is repressed by the insulin-regulated Glycogen Synthase Kinase-3 (GSK3). We have demonstrated that phosphorylation of NRF2 by GSK3 enhances β-TrCP (beta-transducin repeat-containing protein) binding and ubiquitylation by CUL1 (cullin-1), resulting in increased proteasomal degradation of NRF2. Thus, we hypothesise that inhibition of GSK3 activity or β-TrCP binding upregulates NRF2 and so protects beta cells against oxidative stress. We have found that treating the pancreatic beta cell line INS-1 832/13 with the KEAP1 inhibitor TBE31 significantly enhanced NRF2 protein levels. The presence of the GSK3 inhibitor CT99021 or the β-TrCP-NRF2 protein-protein interaction inhibitor PHAR, along with TBE31, resulted in prolonged NRF2 stability and enhanced nuclear localisation (P < 0.05). TBE31-mediated induction of NRF2-target genes encoding NAD(P)H quinone oxidoreductase 1 (Nqo1), glutamate-cysteine ligase modifier (Gclm) subunit and heme oxygenase (Hmox1) was significantly enhanced by the presence of CT99021 or PHAR (P < 0.05) in both INS-1 832/13 and in isolated mouse islets. Identical results were obtained using structurally distinct GSK3 inhibitors and inhibition of KEAP1 with sulforaphane. In summary, we demonstrate that GSK3 and β-TrCP/CUL1 regulate the proteasomal degradation of NRF2, enhancing the impact of KEAP1 regulation, and so contributes to the redox status of pancreatic beta cells. Inhibition of GSK3, or β-TrCP/CUL1 binding to NRF2 may represent a strategy to protect beta cells from oxidative stress.
>展开<收起